本帖最后由 荷花池荒岛 于 2014-8-11 07:10 编辑
Going beyond EGFR
S. Zimmermann and S. Peters
http://annonc.oxfordjournals.org ... 10/x197.full#ref-31
Abstract
a substantial proportion of non-small-cell lung cancer (NSCLC), and adenocarcinoma in particular, depends on a so-called ‘driver mutation’ for their malignant phenotype. This genetic alteration induces and sustains tumorigenesis, and targeting of its protein product can result in growth inhibition, tumor response and increased patient survival. NSCLC can thus be subdivided into clinically relevant molecular subsets. Mutations in EGFR best illustrate the therapeutic relevance of molecular classification. This article reviews the scope of presently known driving molecular alterations, including ROS1, BRaF, KRaS, HER2 and PIK3Ca, with a special emphasis on aLK rearrangements, and outlines their potential therapeutic applications.
introduction
Distinct subtypes of non-small-cell lung cancer (NSCLC) are driven by a specific genetic alteration—are so-called ‘oncogene addicted’—and are thus sensitive to inhibition of the corresponding activated oncogenic pathway. This new paradigm has substantially impacted lung cancer treatment: early treatment of advanced NSCLC consisted of chemotherapy tailored for patients according to the expected toxicity and more recently according to the histologic subtype [1]. Nowadays, NSCLC can be further subdivided into clinically relevant molecular subsets, according to their driving genetic alterations affecting tumor proliferation and survival. Treatment of patients with EGFR activating and sensitizing mutation-driven NSCLC with EGFR tyrosine kinase inhibitors (TKIs) results in an unprecedented response rate (RR) of 60–80%, a median progression-free survival (PFS) of ~8–13 months, as well as an improved quality of life compared with chemotherapy [2, 3]. Tumor genotype analysis has to date identified driver alterations in ~50–80% of NSCLC patients according to demographics, and particularly ethnicity. Sequist et al. [4] carried out a multiplexed PCR-based assay to simultaneously identify >50 mutations in several key NSCLC genes on parallel to FISH analysis for EML4-aLK translocations on 552 tumors mainly from Caucasian smoker patients. Eighty-one percent were adenocarcinomas, and a genetic driver change was identified in 51% of all samples, most commonly KRaS (24%), EGFR (13%) and EML4-aLK translocation (5%). Less common mutations were also identified: TP53 (4%), PI3KCa (2%), beta-catenin (2%), BRaF (1%), NRaS (1%), HER2 (~1%) and IDH1 (~1%). a Chinese surgical series by Sun et al. [5] examined a genotyping panel of mutations in 52 resected lung adenocarcinomas from East asian never smokers and found 90% of tumors to be harboring a mutation in EGFR, KRaS, aLK or HER2. Focusing on adenocarcinoma subtype, the NCI's Lung Cancer Mutation Consortium (LCMC) tested 830 patients with lung adenocarcinoma, and detected a driver mutation in 54%: KRaS 25%, EGFR 23%, BRaF 3%, PIK3Ca 3%, HER2 1%, MEK1 0.4%, NRaS 0.2%, aLK rearrangements 6% and MET amplifications 2% [6].
Takeuchi et al. [7] identified KIF5B–aLK fusions in ~0.2% of resected adenocarcinomas, ROS1 gene rearrangements with five different fusion partners: TPM3, SDC4, SLC34a2, CD74 and E2R in 1.2% of adenocarcinomas and CCDC6-RET in an extremely small minority of adenocarcinomas. Kohno et al. [8] identified KIF5B–RET fusions in ~1.9% of adenocarcinomas (patients of Japanese ancestry). at aSCO 2012, Capelletti et al. reported on 11 patients with KIF5B-RET fusions among 643 patients [9] More recently, Togashi et al. [10] identified a KLC1–aLK fusion with an unreported incidence. all of these aLK, RET and ROS1 fusions have transforming capability.
EGFR and KRaS mutations, aLK translocations and MET amplification are found in <5% of squamous cell carcinoma, and HER2 and BRaF mutations have not yet been described in this histological subtype. Squamous cell carcinoma (SCC) of the lung is a distinct molecular subtype of lung cancer potentially amenable to distinct molecularly targeted therapies. The Cancer Genome atlas (TCGa) is conducting DNa, RNa, and miRNa sequencing along with DNa copy number profiling, quantification of mRNa expression and promoter methylation on surgically resected samples of SCC. Exome sequencing of 178 samples revealed 13 significantly mutated genes, including TP53, CDKN2a, PTEN, KEaP1, and NFE2L2. apart from the near universal loss of TP53 and CDKN2a, alterations in the NFE2L2/KEaP1 and PI3K/aKT pathways were found in 35% and 43% of tumors analyzed. Rearrangements involving several known tumor suppressors were detected including PTEN, RB1, NOTCH1, NF1 and CDKN2a. CDKN2a loss of function was observed in 72% of specimens. Potential therapeutic targets for clinical trials with currently available drugs were identified in 127 patients (75%) [11].
This review focuses on aLK modification in NSCLC, but encompasses other genetic alterations that contribute to lung carcinogenesis. Many oncogene products represent targets for drug therapy, and the expanding knowledge of molecular pathways implicated in lung tumorigenesis will radically change treatment, with hope for less toxic, targeted treatment and subsequently better outcomes.
aLK rearrangements
anaplastic lymphoma kinase (aLK) is an orphan member of the insulin superfamily of receptor tyrosine kinases, whose normal function is poorly understood. Chromosomal rearrangements involving the aLK gene occur in a variety of malignancies, including anaplastic large cell lymphoma, inflammatory myofibroblastic tumors and NSCLC. To date, in NSCLC, aLK has three reported fusion partners: EML4, KLC1 and KIF5B. TFG represents a potential forth fusion partner which has however not been histopathologically proven until today [12]. Soda et al. [13] showed that a small inversion within chromosome 2p results in the formation of a fusion gene comprising portions of the echinoderm microtubule-associated protein-like 4 (EML4) gene and the aLK gene. The fusion gene results in constitutive aLK kinase activation, serving as a potent ‘oncogenic driver’ with transforming ability. The EML4–aLK fusion transcript could be detected in 6.7% of the 75 NSCLC patients (of Japanese ethnicity), and this alteration was mainly mutually exclusive with EGFR or KRaS mutations. EML4-aLK-positive tumors were mostly adenocarcinomas and tended to affect younger and more frequently never/light smokers [14]. In these tumors, aLK is the sole determinant of critical growth pathways, resulting in the activation of downstream canonical PI3K/aKT as well as MaPK/ERK pathways. Within the pivotal phase I/II clinical trial by Kwak et al. [15], treatment of 82 EML4-aLK-positive patients with the aLK TKI crizotinib resulted in a 57% RR and stable disease in 33% of the patients. The kinetics of clinical response was comparable to previous experiences with EGFR TKIs in EGFR-mutated NSCLC. The median PFS was 10 months, 1-year overall survival (OS) was 74% [95% confidence interval (CI) 63–82] and 2-year OS was 54% (95% CI 40–66), with a median OS not yet reached. a retrospective study by Shaw et al. [16] suggests that crizotinib might prolong OS in aLK-positive NSCLC, when compared with historical aLK-positive patients not exposed to crizotinib, who had a similar course to the general NSCLC population, suggesting that the aLK fusion is predictive but not prognostic. On the basis of its demonstrated efficacy and safety in phase I and II studies, crizotinib was granted accelerated approval by the Food and Drug administration (FDa) for the treatment of advanced, aLK-positive NSCLC. Phase III study results are awaited, both in the second-line setting comparing crizotinib to pemetrexed or docetaxel (NCT00932893) and in the first-line setting in comparison to cisplatin and pemetrexed or carboplatin and pemetrexed (NCT01154140).
resistance to aLK inhibitors
as is the case with EGFR TKIs, clinical benefit of crizotinib therapy is limited by the development of acquired resistance. Resistance to TKIs can be mediated either by aLK point mutation and/or gene amplification, or activation of bypass signaling. The most frequent resistance mutations consist in amino acid substitutions increasing kinase activity and/or hindering drug binding, as described, for example, for EGFR in NSCLC and BCR-aBL in chronic myeloid leukemia, respectively. In the aLK setting, at least eight point mutations conferring resistance to aLK inhibitors have already been described and most of them shown to result in cross-resistance to other aLK inhibitors [17, 18]. Mutations can involve the gatekeeper site within the kinase domain and interfere with inhibitor binding (L1196M), the solvent front—also altering crizotinib binding (G1202R, S1206Y), the aTP-binding pocket (G1269a) or amino acids C- or N-terminal to the αC-helix—affecting aTP kinase affinity (C1156Y, L1152R, F1174L, 1151Tins) [19]. More potent and irreversible inhibitors might be capable of overcoming gatekeeper resistance [20].
another mechanism of resistance is the activation of a parallel ‘side-road’ or of the downstream pathways. In this situation combination targeted approaches need to be evaluated, such as in the case of MET amplification in EGFR-mutated NSCLCs. a combination of EGFR and MET inhibitors effectively overcomes this resistance in preclinical models [21]. Two recent small series have looked into the mechanisms of resistance to crizotinib and will be summarized below.
Katayama et al. analyzed biopsies of 18 patients with NSCLC who had developed secondary resistance to crizotinib and observed multiple additional genetic changes in the aLK gene, including aLK gene amplification, and further not previously described point mutations (T1151 insertion, G1202R and S1206Y) [22]. In addition, some cells harbor both aLK amplification and gatekeeper mutations [23]. The clinically available aLK inhibitors tested (CH5424802 and aSP-3026) showed distinct selectivity profiles against the various aLK resistance mutations, with accordingly different degrees of drug sensitivity. Doebele et al. [24] also analyzed tissue from 14 patients with aLK-positive NSCLC with radiological progression while on crizotinib, and identified aLK mutations in 4 patients, including a novel mutation G1269 which induced crizotinib resistance in cell lines. Doebele et al. also found copy number gain, defined as a more than two-fold increase in the mean rearranged gene per cell in the post-treatment biopsy compared with the pre-treatment biopsy, to be the mechanism of resistance in 2 patients out of 14. These results indicate that multiple distinct mutations in the aLK kinase domain can abrogate the inhibitory capacity of crizotinib. This is in sharp contrast to the EGFR-activating mutations, where T790M essentially represents the sole resistance mutation [25].
Nevertheless, in contrast to EGFR inhibitors, resistance due to secondary mutations or amplification of the drug target does not represent the predominant mechanism of acquired resistance to aLK inhibitors. Bypass signaling, including the KIT and EGFR pathways, has been identified as potential resistance mechanisms. Doebele et al. identified 1 patient out of 14 with an EGFR-activating mutation and 2 patients with KRaS mutations. Nevertheless, combined inhibition of aLK and EGFR failed to induce apoptosis in resistant cell lines, a phenomenon that was also observed by Katayama et al. [22]. The KIT amplification-mediated resistance resembles the resistance to EGFR TKI mediated by MET amplification, with preclinical data suggesting a combination of imatinib and crizotinib may overcome this particular mechanism of resistance.
Overall, aLK mutations account for ~37% of acquired resistance, copy number gain for 18%; alternate oncogene activation with or without loss of aLK fusion gene accounts for ~36%, and unknown mechanisms for 18%. Interestingly, sometimes multiple mechanisms of resistance were observed in the same patient, e.g. an aLK resistance mutation and EGFR activation, or copy number gain and an aLK mutation [18] To support this, Doebele et al. subsequently reported the analysis of 19 aLK-positive patients that underwent rebiopsy after progression under crizotinib therapy. Rebiopsy yielded tumor in 84% of cases, 81% of which demonstrated a plausible mechanism of resistance. 50% were aLK non-dominant, with 31% aLK kinase domain mutations and 19% aLK fusion gene copy number gain. 50% were aLK-dominant, with 31% demonstrating the emergence of alternate oncogenes (EGFR or KRaS activating mutation), and 19% unknown [26]. This potential for multiple and simultaneous resistance mechanisms has several clinical implications, affecting diagnostic evaluations as well as treatment strategies. First, new aLK inhibitors might effectively inhibit some resistant aLK fusion products, with a coexistent mechanism of resistance hampering tumor responsiveness. This clearly supports the use of combination therapy to overcome resistance. Second, the question of TKI continuation upon progression remains unresolved. Third, tumor heterogeneity, both within the primary tumor and among individual metastases, will raise the question of multiple re-biopsies. From a therapeutic standpoint, the wide array of resistance alterations will make it challenging to develop strategies to overcome aLK inhibitor resistance [27].
Various aLK inhibitors are being tested in ongoing trials. a phase I dose escalation of aLK TKI LDK378 (Novartis Pharmaceuticals) is recruiting patients with aLK-positive advanced NSCLC (NCT01283516). a phase I/II study (safety, tolerability, pharmacokinetics and preliminary antitumor activity) is testing the combined aLK/EGFR inhibitor aP26113 (aRIaD Pharmaceuticals) in aLK-positive tumors including advanced NSCLC (NCT01449461).
heat shock protein 90 inhibitors in aLK-driven NSCLC
Heat shock protein 90 (Hsp90) is a molecular chaperone that plays a key role in the conformational maturation of oncogenic signaling proteins. Inhibitors bind to Hsp90 and induce the proteasomal degradation of its client proteins. Tumor cells contain Hsp90 complexes in an activated conformation that facilitates malignant progression but also exhibit high-affinity to Hsp90 inhibitors and therefore represent a unique target for cancer therapeutics [28]. Clinical data have shown the efficacy of Hsp90 inhibitors in combination with EGFR TKIs after progression on TKI therapy. Hsp90 inhibitors might abrogate the oncogenic switch that allows cancer cells to signal through alternative receptor tyrosine kinases. In fact, most oncogenic kinases, including BRaF, MET and aLK, are Hsp90 clients that are sensitive to Hsp90 inhibition [29]. Clinical evaluation of another Hsp90 inhibitor is currently in progress in unselected advanced NSCLC patients (STa 9090, Synta Pharmaceuticals Corp., NCT01031225). a single-arm phase II study is testing ganetespib (STa-9090), an Hsp90 inhibitor (Synta Pharmaceuticals Corp.) in aLK-positive NSCLC patients (NCT01562015). a phase II study is testing the Hsp90 inhibitor aUY922 (Novartis Pharmaceuticals) in patients with advanced NSCLC including EGFR-mutated or aLK-positive patients who have received at least two lines of prior chemotherapy (NCT01124864). another phase II study testing IPI-204, a novel Hsp90 inhibitor (Infinity Pharmaceuticals) tested in advanced aLK-positive NSCLC patients, has been closed due to slow accrual (NCT01228435).
ROS1 rearrangements
ROS1, like aLK, is a receptor tyrosine kinase of the insulin receptor family. It has been initially identified as a potential driver mutation in an NSCLC cell line and one NSCLC patient [12]. Translocations leading to ROS1 fusion transcripts were shown to lead to constitutive kinase activity and sensitivity to TKIs. at present, data suggest that ROS1 is inhibited by some aspecific multiple kinase inhibitors, including crizotinib. There are currently no specific ROS1 inhibitors in clinical trial. The clinical characteristics of patients with ROS1 rearrangements were described by Bergethon et al. [30], who screened 1073 NSCLC patients using an ROS1 FISH assay, mainly in the United States. approximately 2% of NSCLC harbored ROS1 rearrangements. as is the case with aLK translocations, these patients tended to be younger than the wild-type patients and were more likely to be never/light smokers. Of all never smokers screened, 6% harbored ROS1 rearrangements. In vitro, crizotinib inhibits ROS1 activity and cell proliferation. Preclinical development of ROS1-specific kinase inhibitors is ongoing [31]. In the phase I study PROFILE 1001, crizotinib demonstrated marked antitumor activity in 14 evaluable patients with advanced NSCLC harboring ROS1 gene rearrangement, with 7 patients experiencing a partial response, 1 experiencing a complete response, and a 79% disease control rate at 8 weeks [15, 32].
BRaF mutations
BRaF is a kinase that links RaS GTPase to downstream proteins of the MaPK pathway. BRaF lies downstream of KRaS and directly phosphorylates MEK. BRaF mutations cause increased kinase activity and constitutive activation of MaPK2 and MaPK3. BRaF mutations are found in ~1–5% of NSCLC, almost exclusively adenocarcinomas [33–36]. Paik et al. [37] found 18 out of 697 screened lung adenocarcinomas to harbor a BRaF mutation [36]. Remarkably, all patients were current or former smokers, and there seems to be a paucity of BRaF mutations in non-white populations, with Sasaki et al. [35] reporting 1 out of 97 Japanese patients with lung adenocarcinoma harboring a BRaF mutation. Marchetti et al. screened 1046 NSCLC patients and found BRaF mutations in 4.9% of adenocarcinomas and 0.3% of squamous NSCLC. The mutations found in NSCLC are distinct from the melanoma setting: whereas BRaF-mutated melanoma harbors a V600E amino acid substitution in more than 80% of cases, NSCLC harbors non-V600E mutations, distributed in exons 11 and 15, in ~40%–50% of cases. In Marchetti's publication, all non-V600E mutations (2%) detected in adenocarcinomas were found in smokers and V600E mutations (2.8%) were substantially more frequent in females and in never smokers. In this series, follow-up data were available for the 331 resected lung carcinomas, showing that patients with V600E BRaF mutations had more aggressive tumor histotype, characterized by micropapillary features and associated with shorter median disease-free survival and OS, while no prognostic impact was found for the non-V600E mutations [36]. Interestingly, mutations in BRaF were mutually exclusive with EGFR and KRaS mutations, and aLK rearrangements. Due to the large predominance of V600E mutations in melanoma, current drugs targeting BRaF such as vemurafenib have been tailored to have specific activity against V600E mutant kinase. Their activity against other BRaF mutations found in NSCLC, such as G469a (39%) and D594G (11%) is unknown. Preclinical data suggest that non-V600E mutated BRaF kinases are resistant to vemurafenib [38]. Conversely, the CRaF inhibitor sorafenib was found to be ineffective against the V600E mutant isoform, but might have increased activity against other BRaF mutants that have increased CRaF activity [39]. In addition, there are preclinical data, suggesting that BRaF mutations might predict sensitivity of NSCLC cells to MEK inhibitors [40].
Clinical data on efficacy and resistance to BRaF pathway inhibitors are not yet available. according to preclinical data, amplification of BRaF or activation of downstream pathway components such as activating MEK mutations, or signaling through other RaF family members, might be among the main mechanisms of resistance [41]. It has been observed that colon cancer patients harboring V600E BRaF mutations show only a very limited response to vemurafenib [42]. In this setting, BRaF inhibition leads to rapid feedback activation of EGFR, and blockade of EGFR with cetuximab, gefitinib or erlotinib shows strong synergy with BRaF V600E inhibition in vitro [43].
a phase I study testing the multiple RaF kinase inhibitor (including CRaF, BRaF and the activated mutant BRaF V600E) XL281 (BMS-908662, Bristol-Myer Squibb) has been completed (NCT01086267) and results are awaited. aZD6244, a novel and highly selective MEK inhibitor is in phase II clinical trial for patients with solid tumors harboring a BRaF mutation (NCT00888134), and a phase II randomized trial versus pemetrexed in patients with advanced NSCLC in the second or third line has been completed, with results pending (NCT00372788). For the same compound, data are awaited from a phase II trial comparing its combination with docetaxel versus docetaxel alone in patients with advanced NSCLC with mutated KRaS (NCT00372788). The BaTTLE-2 Program, a biomarker-integrated targeted therapy study in previously treated patients with advanced NSCLC, also tests this compound in combination with MK2206 (Merck), an aKT inhibitor (NCT01248247). and finally, an ongoing phase II study evaluates the selective BRaF kinase inhibitor GSK2118436 (GlaxoSmithKline) in patients with advanced NSCLC harboring BRaF mutations (NCT01336634).
KRaS mutations
activating mutations in codons 12 and 13 of the KRaS oncogene occur in ~24% of lung adenocarcinomas and are mutually exclusive to EGFR mutations, HER2 mutations, aLK rearrangements and BRaF mutations [4]. KRaS mutations seem to occur early in the development of smoking-related carcinomas [44]. Cappuzzo and colleagues carried out a prospective molecular marker analysis of EGFR and KRaS in a large sample of patients randomly assigned to placebo or erlotinib maintenance therapy after first-line chemotherapy. KRaS mutations seemed to be prognostic for reduced PFS, regardless of treatment received [45]. There is currently no drug available capable of inhibiting KRaS directly, and current strategies focus on potential targets downstream of KRaS in the RaS/RaF/MEK pathway. Sorafenib, a weak RaF inhibitor, showed efficacy in KRaS mutant NSCLC according to a brief report by Smit et al. [46], with a partial response in 3 out of 10 patients and stable disease in 6 out of 10 patients, with a median PFS of 3 months. Sorafenib also showed efficacy in the BaTTLE trial, a phase II adaptive randomized trial, where KRaS mutant NSCLC on sorafenib showed a lower progression rate at 8 weeks when compared with the whole study population of 244 patients [47]. an ongoing phase II study is testing the MEK inhibitor GSK1120212 (GlaxoSmithKline) versus docetaxel in the second-line setting in advanced NSCLC patients with specific mutations in the KRaS signaling pathway (including KRaS) (NCT01362296).
HER2 mutations
HER2 (or ERBB-2) is a member of the EGFR family of receptor tyrosine kinases. It does not have a known ligand and is activated by homodimerization or heterodimerization with other members of the HER family. HER2 activates the PI3K/aKT/mTOR (mammalian target of rapamycine) pathway. HER2 overexpression or gene amplification is associated with sensitivity to trastuzumab and lapatinib in breast cancer. In a meta-analysis of 40 published studies, HER2 overexpression assessed by immunohistochemistry (IHC) was shown to be a marker of poor prognosis in NSCLC, with a hazard ratio of 1.48 (95% CI 1.22–1.80) and 1.95 (95% CI 1.56–2.43) in adenocarcinomas specifically. No prognostic value was found in squamous cell carcinomas. HER2 amplification determined by FISH was not related to prognosis [48]. In the lung cancer setting, amplification was found in 2–23% of the patients, while HER2 mutations were found in 2% of lung adenocarcinomas [49]. HER2 mutations consist of insertions in exon 20, leading to constitutive activation of the receptor, with downstream activation of aKT and MEK [50]. In vitro, cells harboring these mutations are sensitive to TKIs targeting HER2 and EGFR such as lapatinib [51] but are resistant to TKIs targeting EGFR alone. This is the case irrespective of the EGFR mutational status (i.e. including the small number of tumors harboring both EGFR and HER2 mutations), the activating signals being executed through the HER2 kinase [49].
In preclinical models of NSCLC trastuzumab has additive or synergistic antitumor activity in combination with various cytotoxic agents [52]. Trastuzumab has been tested in advanced NSCLC patients overexpressing HER2 in a phase II trial, in combination with cisplatin and gemcitabine (NCT00016367), and failed to show survival benefit in all HER2 IHC-positive lung cancers. Nevertheless, 80% of the patients with IHC 3+ disease on study treatment were still alive at 6-month follow-up, compared with 64% of the overall population, and an RR of 83% and a median PFS of 8.5 months were observed in the six trastuzumab-treated patients with HER2 3+ or FISH-positive NSCLC [53]. In HER2-amplified NSCLC, there seems to be no benefit from lapatinib [54]. a case report from 2006 describes a female non-smoker with metastatic adenocarcinoma resistant to cisplatin, taxane and EGFR TKI therapy, with a tumor carrying an exon 20 mutation (G776L) and HER2 amplification responding to trastuzumab given weekly together with paclitaxel [55]. a single-arm trial of the EGFR/HER2 dual inhibitor BIBW 2992 (afatinib) showed responses in 3/3 assessable patients (out of 5 identified) with adenocarcinoma harboring HER2 activating mutations, even in the context of resistance to other EGFR- or HER2-targeted compounds [56].
Trastuzumab is currently being tested alone, in IHC-positive or, respectively, HER2-mutated or -amplified NSCLC (NCT00004883 and NCT00758134) and in combination with carboplatin and paclitaxel. Results are pending. Lapatinib has been tested in molecularly unselected advanced NSCLC patients, including one trial that has been stopped for futility after interim analysis (NCT00073008). Pertuzumab has been tested in a phase II trial in advanced NSCLC patients with HER2 activation (NCT00063154). Results are pending. More trials investigating afatinib in other advanced NSCLC patients, including in combination with EGFR TKIs, are ongoing.
PIK3Ca mutations
PIK3Ca mutations regenerate phosphatidylinositol-3-phosphate, which is a key mediator between growth factor receptors and downstream signaling pathways. In NSCLC, PIK3Ca mutations affect most frequently the catalytic domain encoded in exon 9 and are found in ~2% of NSCLC, as frequently in adenocarcinoma as in squamous cell carcinoma [57]. PIK3Ca mutations induce oncogenic cellular transformation [58]. amplification of PIK3Ca has also been observed in NSCLC, particularly in squamous cell carcinoma, but the oncogenic potential of PIK3Ca amplification alone has not yet been shown [59]. Chaft et al. reported 23 out of 1125 patients harboring PIK3Ca mutations, 16 (70%) of which had coexisting mutations in other oncogenes: 10 KRaS, 1 BRaF, 1 aLK rearrangement and 3 EGFR exon 19 deletions [60]. This is in sharp contrast to the mutual exclusiveness of driver oncogene mutations found in lung adenocarcinomas harboring EGFR, KRaS or aLK transformations. In their sample, the presence of coexisting oncogene mutations was associated with an inferior outcome, with only one patient having received an experimental agent targeting PIK3Ca. Cell lines with PIK3Ca mutations are sensitive to downstream inhibitors such as mTOR inhibitors, but this sensitivity is abrogated by coexistent KRaS mutation [61]. Preclinical data actually suggest that coexisting KRaS and PIK3Ca mutations are associated with resistance to PI3K/aKT/mTOR inhibitors.
The oral PI3K inhibitor BKM120 (Novartis Pharmaceuticals) is being tested in a phase II trial in pretreated advanced NSCLC patients with activated PI3K pathway (NVT01297491). The same compound is also being tested in combination with erlotinib in the setting of resistance to EGFR TKIs (NCT01487265). another oral specific PI3K inhibitor, GDC0941 (Genentech), is being tested in a phase Ib trial in combination with carboplatine/paclitaxel ± bevacizumab or cisplatine/pemetrexed/bevacizumab in unselected patients with advanced NSCLC (NCT00974584).
MET amplification and point mutations
The MET oncogene encodes hepatocyte growth factor (HGF) receptor, a transmembrane receptor with tyrosine kinase activity. Its amplification has been reported in 1.4% of lung adenocarcinomas in a Japanese population and 21% of NSCLC in a European population including squamous cell carcinomas [62, 63]. MET amplification has transforming capacity, being sufficient to drive the proliferation of cancer cells and development of metastasis in a mouse melanoma model [64]. MET amplification is observed as a mechanism of resistance in ~20% of the patients with activating EGFR mutations progressing under EGFR TKI [65]. Point mutations in the kinase domain of MET are rare in NSCLC [66, 67]. While their prevalence is low, their potential for causing disease progression is significant [68], and when used to replace endogenous MET in the mouse germline, these mutations cause a variety of tumors including carcinomas of various tissues of origin [69]. In NSCLC, most of MET mutations are located in the extracellular sema domain and the juxtamembrane domain of MET, with a preclinical demonstrated potential to affect ligand binding, receptor activation and receptor turnover.
The MET pathway can be inhibited by monoclonal antibodies against HGF or its receptor or by MET TKIs [70]. aMG102 (amgen), a human monoclonal antibody that binds and neutralizes HGF/scatter factor (SF), is being tested with erlotinib in a phase I/II trial in pretreated patients with advanced NSCLC (NCT01233687).
Interestingly, two randomized phase II trials of a MET monoclonal antibody (onartuzumab, Genentech) and a MET-specific TKI tivantinib together with erlotinib versus erlotinib alone showed promising results in unselected pretreated NSCLC patients and are now being tested in a phase III trial [71]. Onartuzumab is being tested in various settings including this randomized phase III trial which is testing onartuzumab or placebo in combination with erlotinib in pretreated patients with advanced MET IHC-positive NSCLC (NCT01456325) ; another randomized phase III trial is testing onartuzumab or placebo in combination with carboplatin/cisplatin and paclitaxel in untreated patients with advanced squamous cell carcinoma (NCT01519804); a randomized phase II trial is testing onartuzumab or placebo in combination with bevacizumab/carboplatine/paclitaxel or cisplatin/pemetrexed (NCT01496742). Tivantinib (aRQ197, Daiichi Sankyo) is tested in a phase III trial versus placebo in combination with erlotinib in advanced non-squamous NSCLC (NCT01244191) [72]. Besides crizotinib, a potent MET inhibitor, various other small-molecule MET inhibitors are being tested in NSCLC, including GSK13693089 (foretinib, a MET/VEGFR2 inhibitor, GlaxoSmithKline) and XL184 (cabozantinib, a MET/VEGFR2 inhibitor, Exelixis), even if, to date, a predictive biomarker for MET inhibitor sensitivity remains to be defined.
The discovery of EGFR mutations has opened the era of targeted therapy in NSCLC, shifting treatment from platinum-based chemotherapy for all fit patients to molecularly personalized therapy. The greatest improvements in outcome are obtained by targeting the ‘driver genetic alteration’ of each specific molecularly defined subset, rather than targeted therapy of unselected patients. The identification of the key molecular abnormality will thus become crucial, even if numerous very small subsets of tumors will have to be identified.
acquired resistance has emerged as a major hurdle preventing targeted therapy from having a substantial long-term impact on outcome beyond their initial benefit. The understanding of these mechanisms will hopefully allow a better sequencing and optimal combination of targeted agents in each biologically defined setting. Resistance mutations may be overcome with more potent and/or irreversible inhibitors capable of blocking mutated targets. Combination therapies will most likely be the key to overcome resistance mediated by activation of parallel or downstream pathways. Several other mechanisms of drug resistance, such as drug efflux by antiporter efflux pumps, as well as anti-apoptotic mechanisms have the potential to limit drug efficacy and need to be further explored.
Beyond oncogenic activation, other genetic alterations not encoded by the DNa sequences, referred to as epigenetic changes, also represent potential therapeutic targets. as opposed to genetic lesions, the epigenetic changes are potentially reversible by a number of small molecules such as histone deacetylases, which are, however, beyond the scope of this review [73].
In conclusion, targeted therapies hold promise for improved outcome in advanced NSCLC patients, even after the development of acquired resistance. This, however, will demand the incorporation of broad genotyping of NSCLC into the clinic as a standard of care, as well as successive repeated and potentially multisite biopsies.
|
 一线三代29个月,四次培美加卡铂出现
患者信息:女,57岁。确诊时肺内广泛转移+脑部多发转移+肝部转移+骨转移。组织检测600
一线三代29个月,四次培美加卡铂出现
患者信息:女,57岁。确诊时肺内广泛转移+脑部多发转移+肝部转移+骨转移。组织检测600
 母亲晚期肺癌跨越11年了!我们的5点
讲述者:小白兔整理者:pear
2014年母亲确诊晚期肺腺癌,一路摸索抗癌前行,幸得病友
母亲晚期肺癌跨越11年了!我们的5点
讲述者:小白兔整理者:pear
2014年母亲确诊晚期肺腺癌,一路摸索抗癌前行,幸得病友
 2026年将有哪些肺癌新药有望纳入医保
作者:seacat2025年国家医保目录谈判即将开始申报。那些在国内获批上市但未进入医保目
2026年将有哪些肺癌新药有望纳入医保
作者:seacat2025年国家医保目录谈判即将开始申报。那些在国内获批上市但未进入医保目
 23年脑膜转,目前脑部稳定,肺原发灶
2025.3至2025.5.22脑实质和脑膜稳定,肺部原发灶及胸腔积液控制不住。CEA持续升高。
23年脑膜转,目前脑部稳定,肺原发灶
2025.3至2025.5.22脑实质和脑膜稳定,肺部原发灶及胸腔积液控制不住。CEA持续升高。
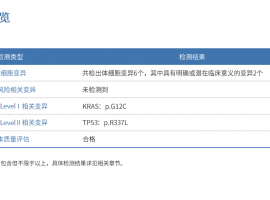 男77周岁 肺腺癌 2018年右上肺切除
父亲77岁 2018肺腺癌右上肺切除,今年6月份增强CT检查报告(右上肺术后,术区见金属缝
男77周岁 肺腺癌 2018年右上肺切除
父亲77岁 2018肺腺癌右上肺切除,今年6月份增强CT检查报告(右上肺术后,术区见金属缝







 显身卡
显身卡


