本帖最后由 漓江渔翁 于 2015-7-23 15:26 编辑
非小细胞肺癌对易瑞沙耐药的作用机制
时间:2014-12-08 08:36来源:51奇迹 作者:网络 点击: 312 次
非小细胞肺癌对易瑞沙耐药的作用机制涉及多种基因水平改变和信号转导通路异常激活。HSP90是广泛存在于生物体内的多功能分子伴侣,它的“客户蛋白”大多是肿瘤细胞增殖、分化、侵袭、转移及血管生成等重要过程所依赖的蛋白激酶、转录因子或癌基因的产物,包括突变的EGFR、血管内皮生长因子受体(vascular endothelial cell growth factor receptor, VEGFR)、人表皮生长因子受体2(human epidermal growth factor 2, HER2)、survivin、缺氧诱导因子-1α(hypoxia inducible factor-1, HIF-1α)及细胞周期调节因子(CDK4、CDK6)等[7]。抑制HSP90可导致这些“客户蛋白”通过泛素-蛋白酶体途径发生降解,从多种途径和环节产生抗肿瘤作用而实现多靶点攻击。
本研究中,A549细胞株为野生型EGFR,同时含有K-ras基因突变。约10%-30%的非小细胞肺癌患者存在K-ras基因突变,它是导致易瑞沙原发性耐药的重要机制。Pao等[8]用TKI治疗60例肺腺癌患者时发现38例不敏感的患者有9例存在K-ras基因突变,Massarelli等[9]认为K-ras突变和EGFR突变相互排斥,是评价非小细胞肺癌患者能否接受TKI治疗的阴性预测因子。H1975细胞株的EGFR含有L858R和T790M双突变。L858R为EGFR第21外显子858位点亮氨酸置换为精氨酸的点突变,是TKI治疗的敏感突变。T790M是发生在EGFR第20外显子790位密码子苏氨酸置换为甲硫氨酸的错义突变,非小细胞肺癌对易瑞沙的获得性耐药50%以上是由T790M突变造成的[10-12],它是国际公认的重要获得性耐药机制。
我们应用体外实验的方法, 将HSP90抑制剂17-DMAG作用于两种遗传背景不同的非小细胞肺癌细胞株A549和H1975,结果表明它对两种细胞均产生了明显的增殖抑制作用,与Shimamura等的研究结果相似,而Kobayashi等[6]则认为17-DMAG不能抑制A549细胞增殖,可能与本实验药物作用剂量梯度不同有关。A549细胞的K-ras突变直接持续激活Ras-Raf-MEK-MAPK信号通路,并失去对上游EGFR的依赖性,导致TKI耐药的产生,而H1975细胞依赖EGFR突变后异常激活的多条下游信号转导通路维持生存和增殖,HSP90抑制剂能同时阻断这些信号通路从而抑制细胞增殖。
本研究显示17-DMAG能促进A549和H1975细胞凋亡,作用48 h后的平均凋亡率分别为11.73%和16.88%,造成总体凋亡率较低的原因可能与细胞状态、处理过程及检测方法等因素有关。研究表明,细胞周期蛋白依赖性蛋白激酶家族的CDK4和CDK6是HSP90的“客户蛋白”,抑制HSP90可以降低它们的表达水平,使细胞出现生长周期阻滞并发生凋亡。Survivin是目前已知最强的凋亡抑制因子,在多种恶性肿瘤中呈高表达状态,抑制HSP90能使它发生降解,从而促进肿瘤细胞凋亡。这些都可能是17-DMAG作用后细胞发生凋亡的原因。此外,我们发现17-DMAG对两种细胞HSP90的表达均无明显影响,再次证实它只是发挥分子伴侣的作用,其本身在药物作用之后并不会发生降解。
相关研究认为,HSP90抑制剂可下调A549细胞先前存在或重新激活的HER2及PI3K/Akt和STAT3/STAT5信号级联反应中的关键调节因子的表达,而与野生型EGFR的存在无关,这可能是EGFR在17-DMAG作用后未发生明显降解的原因。而H1975细胞中突变的EGFR需要HSP90来维持它的构象成熟和状态稳定以保证正常功能的发挥,所以对于HSP90抑制剂引起的降解效应更为敏感。相关的具体作用机制尚有待于进一步证实。
近年来,HSP90抑制剂作为多靶点的抗肿瘤药物因其独特的优越性已在各种恶性肿瘤治疗方面进行了不同时期的临床试验。随着肺癌的发病机制不断阐明,根据患者的自身情况和基因状态设计合理的用药方案从而实现真正的个体化治疗是未来肺癌临床研究的前沿和热点,HSP90抑制剂也将越来越受到人们的关注并发挥重要作用。
|
 三代伏美换一代特罗凯获益率问题
家父25年2月因肩膀疼痛确诊肺腺癌晚期4B,双肺转,多发骨转,脑转(非典型),
三代伏美换一代特罗凯获益率问题
家父25年2月因肩膀疼痛确诊肺腺癌晚期4B,双肺转,多发骨转,脑转(非典型),
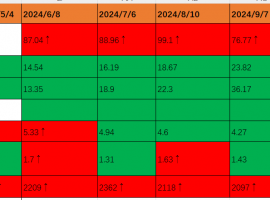 盲试靶向药 29个月,治疗分享
时间:2025/1/27 盲试靶向药第29个月。
本月治疗方案:7080(14mg)。2024年11月底
盲试靶向药 29个月,治疗分享
时间:2025/1/27 盲试靶向药第29个月。
本月治疗方案:7080(14mg)。2024年11月底
 奥西替尼耐药后续治疗,出现腹水
治疗经过:
2010确诊腺癌
治疗方案:右肺上叶切除,10年民诺宾2次,顺铂+盖诺2次。
奥西替尼耐药后续治疗,出现腹水
治疗经过:
2010确诊腺癌
治疗方案:右肺上叶切除,10年民诺宾2次,顺铂+盖诺2次。
 完成手术,病理也出来了,烦请各位老
之前因为心脏问题,去心内造影,前降支75%狭窄,回到心胸外科手术,目前主治医生说是
完成手术,病理也出来了,烦请各位老
之前因为心脏问题,去心内造影,前降支75%狭窄,回到心胸外科手术,目前主治医生说是
 生存期超4年!首个击败奥希替尼的药
作者:seacat
2025年3月26日,巴黎举行的2025欧洲肺癌大会(ELCC 2025)报道了III期研
生存期超4年!首个击败奥希替尼的药
作者:seacat
2025年3月26日,巴黎举行的2025欧洲肺癌大会(ELCC 2025)报道了III期研





 显身卡
显身卡 3IK突变或者扩增)
3IK突变或者扩增)


